




-
As a result of globalisation and industrialisation, the concentration of carbon dioxide (CO2) in the atmosphere has increased significantly in recent years1, drawing attention to the urgent need for effective approaches to reduce CO2 emissions. When produced through sustainable processes, hydrocarbons derived from CO2 can serve as carbon-neutral fuels, enabling their seamless integration into the existing energy infrastructure, thereby reducing the reliance on non-renewable fossil fuels2.
One important technique that has a lot of potential for solving this problem is catalysis. Metal oxides (TiO2, Cu2O, Co3O4, etc.)3–5, 2D materials (MXene, graphene oxide, MoS2, etc.)6–8, and metal-organic frameworks (MOFs)9–11 are among the many catalysts that have been studied for CO2 conversion. Despite their effectiveness, traditional catalysts have significant disadvantages such as poor visible light-harvesting capability, insufficient charge potential for reactions, and low selectivity10,12. These challenges highlight the need for innovative catalytic strategies to improve the effectiveness and scalability of the CO2 reduction reactions.
In recent years, halide perovskite-based catalysts have attracted considerable interest because of their unique properties, including tunable band gap, high charge carrier mobility for efficient charge separation, strong light absorption, and high surface area for perovskites in the form of nanocrystals (NCs)13–17. Owing to these properties, perovskites have demonstrated considerable potential in photocatalysis (PC), in which visible light is used to drive selective chemical reactions, particularly CO2 reduction, for efficient conversion into valuable products such as methane and formic acid18. Moreover, they are particularly suitable for solar-driven catalysis because sunlight is a readily available renewable energy source.
For example, the incorporation of halide perovskites significantly enhances the catalytic properties of TiO2. Specifically, hybrid TiO2/CsPbBr3 nanofibres have demonstrated superior performance in photocatalytic CO2 reduction, achieving a higher production rate of 9.02 μmol·g−1·h−1 compared to 4.68 μmol·g−1·h−1 for pristine TiO2 nanofibres, thereby confirming the enhanced catalytic activity18. In another study, a more complex heterostructured CsPbBr3/Au/TiO2 catalyst showed a 5.4-fold increase in performance in comparison to the CsPbBr3/TiO2 structure due to the formation of an ohmic contact, enabling fast charge transfer across the CsPbBr3 and TiO2 interface19. Au@CdS/CsPbBr3 catalyst was also employed for the CO2 photoreduction reaction, achieving a rate of approximately 3 μmol·g−1·h−1 with an apparent quantum yield of about 0.27%, where CdS served as a mediator layer between the CsPbBr3 and gold (Au) plasmonic antennas, which generate hot electrons20. Thus, the usage of lead-halide perovskites provides an improvement over traditional catalysts. However, these studies have some limitations, such as selectivity because the main products of PC are hydrogen (H2), carbon monoxide (CO), and methane (CH4). In this regard, several studies have demonstrated that the application of protective shell coatings is an effective strategy for enhancing the stability of halide perovskites in various applications21–23. Nevertheless, achieving a long-term stable photocatalytic performance using perovskite-based nanomaterials remains a challenge.
In this study, we develop water-stable and highly selective photoelectrocatalytic materials for efficient CO2 reduction reactions. For this purpose, we modify the hot-injection protocol for the synthesis of core-shell CsPbBr3@TiO2 heterostructures, which are stable not only in polar liquids but also in water. The CsPbBr3@TiO2 heterostructures allow us to achieve high photocatalytic activity for the conversion of CO2 to CH4. Subsequently, we show that the catalytic performance of CsPbBr3@TiO2 perovskite heterostructures can be enhanced by designing CsPbBr3@TiO2 core-shell nanoparticles (NPs) deposited on a gold nanoparticle (NP) layer, leveraging the phenomena of localised surface plasmon resonance (LSPR) and the generation of hot carriers. Thus, we achieve the conversion of CO2 into valuable products, such as ethylene (C2H4) and propylene (C3H6), with high selectivity. Water is used as the reaction medium to facilitate the catalytic process, which significantly reduce the overall cost by eliminating the need for expensive supporting electrolytes and solvents. These findings indicate that perovskite-based nanomaterials are suitable for large-scale and sustainable CO2 utilisation and conversion into fuel.
-
In this study, we present a novel approach to CO2 conversion by combining the advantages of perovskite-based PEC catalysis in water media with LSPR enhancement. By addressing the problem of lead halide perovskite stability in water, we establish a foundation for the development of a robust and scalable photoelectrocatalytic system for efficient CO2 reduction. In this study, we aimed to control the selectivity of the CO2 reduction reaction using various processes (electrocatalytic, photocatalytic, and photoelectrocatalytic) in perovskite-based heterostructures. To achieve this, we synthesised CsPbBr3@TiO2 core NPs and investigated their structures and optical properties. We then prepared the catalysts by stepwise impregnation of copper foam with colloidal gold and CsPbBr3@TiO2 core-shell NPs.
For comprehensive structural analysis, we employed techniques such as transmission electron microscopy (TEM), scanning electron microscopy (SEM), X-ray photoelectron spectroscopy (XPS), and X-ray diffraction (XRD). Their optical properties were studied using absorption and photoluminescence (PL) spectroscopy. Time-resolved photoluminescence (TRPL), electron paramagnetic resonance (EPR), electrochemical impedance spectroscopy (EIS), and transient photocurrent (TPC) measurements were performed to analyse the charge separation in the CsPbBr3@TiO2 and CsPbBr3@TiO2/Au heterostructures. Gas chromatography was used to analyse the products of the CO2 reduction reaction and the stability of the catalysts was studied using chronoamperometry. We present experimental data showing how we can control the selectivity of the CO2 reduction reaction to form H2, CO, CH4, C2H4, or C3H6. To provide a deeper understanding of these processes, we discuss possible reaction pathways for CO2 reduction based on the experimental results.
For the synthesis (Fig. 1) of the CsPbBr3@TiO2 core-shell NPs, we modified the standard hot-injection protocol24. First, PbBr2 was mixed with the 1-octadecene (ODE) and dried under vacuum. Subsequently, oleic acid (OA) and oleylamine (OAm) were added to the PbBr2 suspension in the ODE until the solution became clear. Then, Cs-oleate was injected into the flask, and TBT and water, pre-mixed in ODE, were added stepwise. After 10 min, the reaction mixture was cooled, and the CsPbBr3@TiO2 core-shell NPs were purified.
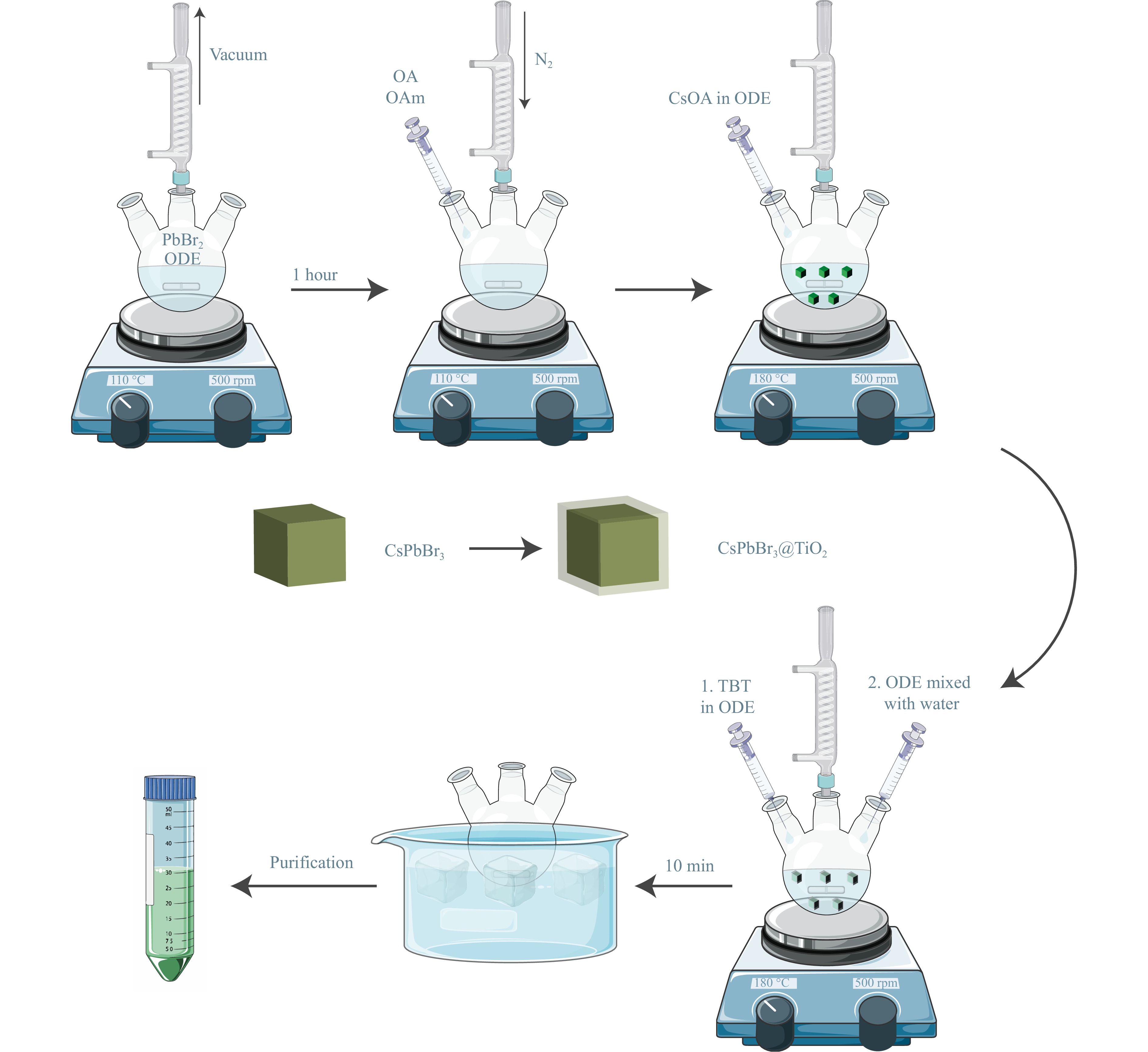
Fig. 1 Scheme of modified hot-injection synthesis of CsPbBr3@TiO2 NPs.
The resulting CsPbBr3@TiO2 core-shell NPs are shown in the HRTEM images (Fig. 2a, c, d). The elemental composition, including Cs, Pb, Br, and Ti (Fig. 2b), was determined using EDX mapping. Interplanar distances corresponding to 4.2 Å (100) were defined using fast Fourier transform (FFT) analysis of the HRTEM images (Fig. 2c, d). Perovskite NCs are known to form after the injection of Cs-oleate during synthesis. In our modified protocol (Fig. 2), TBT was first added after Cs-oleate injection, followed by the addition of water to initiate the hydrolysis reaction, resulting in the coating of the particles. Based on this approach, we conclude that the CsPbBr3 NCs were successfully covered with TiO2 shells. The presence of the shell is supported by the contrast difference and amorphous halo observed in the HRTEM images (Fig. 2a, c, d) as well as by EDX mapping showing the presence of Ti (Fig. 2b). The XRD diffractograms of the CsPbBr3 and CsPbBr3@TiO2 core-shell NPs exhibited strong diffraction peaks corresponding to the (100), (110), (111), (200), (210), (211), (220), and (300) planes of the cubic perovskite phase. The thin TiO2 shell complicates definitive structural determination using HRTEM alone. In the XRD pattern obtained for the CsPbBr3@TiO2 system (Fig. 2e), additional weak reflections are marked with stars. While the peaks observed at ~25° and ~27° may superficially resemble those of the anatase and rutile phases, their assignment to crystalline TiO2 is unlikely, considering the low-temperature, ambient conditions used during shell formation. Instead, these peaks precisely match the reference pattern of the Cs4PbBr6 phase (PDF-2 ICDD card No. 01-073-2478), which is a known secondary phase that may form through the partial degradation of CsPbBr3 under the processing conditions.
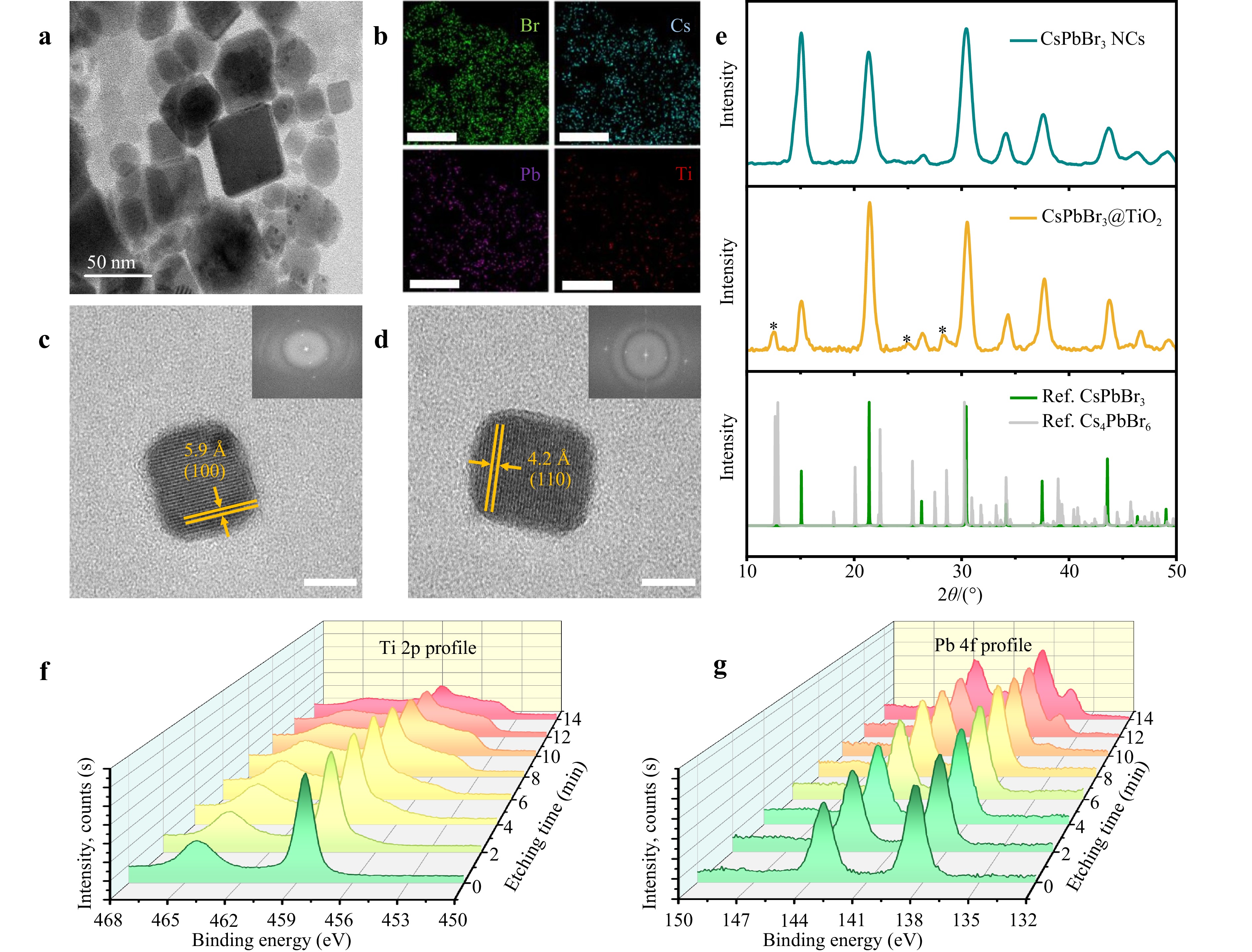
Fig. 2 Structural characterization of CsPbBr3@TiO2 NPs. a TEM image of CsPbBr3@TiO2; b HAADF-STEM and STEM-EDX mapping of Br, Cs, Pb, Ti; c-d HRTEM images of individual CsPbBr3@TiO2 NPs with the corresponding fast Fourier transformation images; e XRD patterns of pristine CsPbBr3 NCs and CsPbBr3@TiO2 NPs. Scale bars correspond to 50 nm (a,b); 10 nm (c,d); f XPS Depth profiling of Ti 2p region; g XPS depth profiling of Pb 4f region.
To further evaluate the presence and integrity of the TiO2 shell, we conducted controlled Ar+-ion sputtering combined with high-resolution XPS measurements of the Ti 2p and Pb 4f core levels (Fig. 2f, g). The sputtering rate was adjusted to approximately 0.5 nm/min to enable gradual removal of surface layers without immediate disruption of the core. During the initial stages of sputtering, the Ti 2p spectra (Fig. 2f) exhibited progressive changes in the peak shape and the appearance of lower binding energy shoulders corresponding to Ti3+ and Ti2+ species, indicating partial reduction of the shell under ion bombardment. In contrast, the Pb 4f spectra (Fig. 2g) remained unchanged in shape and position, whereas their intensity increased with etching time, suggesting that the perovskite core was still chemically intact and protected by the TiO2 layer. Subtle asymmetry in the Pb 4f line appeared only after an estimated etching depth of ~4 nm, and a distinct shoulder at ~137 eV, indicative of Pb0 formation, emerged beyond ~7 nm of sputtering. These results are in excellent agreement with the HRTEM data showing a TiO2 shell thickness of 4–5 nm, and they confirm that the outer shell effectively prevents the immediate degradation of the perovskite during ion exposure. This experiment provides complementary evidence for the core–shell architecture and demonstrates the utility of differential XPS profiling as a non-trivial but informative diagnostic tool for heterostructured nanocomposites.
The survey spectra (Fig. 3g) identified the composition of freshly synthesised CsPbBr3@TiO2 core-shell NPs and CsPbBr3@TiO2/Au heterostructures, as well as control samples of CsPbBr3 NСs and TiO2 synthesised under identical conditions. High-resolution spectra of O1s, Ti2p, Cs3d, Pb4f, Br3d, and Au4f were acquired (Figure 3a-f) to refine the chemical state analysis. The high-resolution spectra of Cs3d5 (724 eV), Pb4f7 (138.1 eV), and Br3d5 (68 eV) for the control CsPbBr3 NСs (Fig. 3a-c) revealed chemical states of Cs+, Pb2+, and Br−, consistent with reported data for bromide perovskites25. The binding energies of O1s (530 eV) and Ti2p3 (458.53 eV) for the TiO2 control sample (Fig. 4d, e) confirmed the oxidation states of O2− and Ti4+, supporting the formation of titanium dioxide26. Quantitative XPS analysis, presented in Table S1 indicates that the stoichiometric ratio for perovskite is Cs:Pb:Br at approximately 1:1:3, and that for TiO2, Ti:O is 1:2. Comparisons of the high-resolution spectra (Fig. 4a-c) for the CsPbBr3@TiO2 and CsPbBr3@TiO2/Au structures with control samples revealed shifts and changes in the emission line shapes. The peak positions and FWHM values are detailed in Table S2.
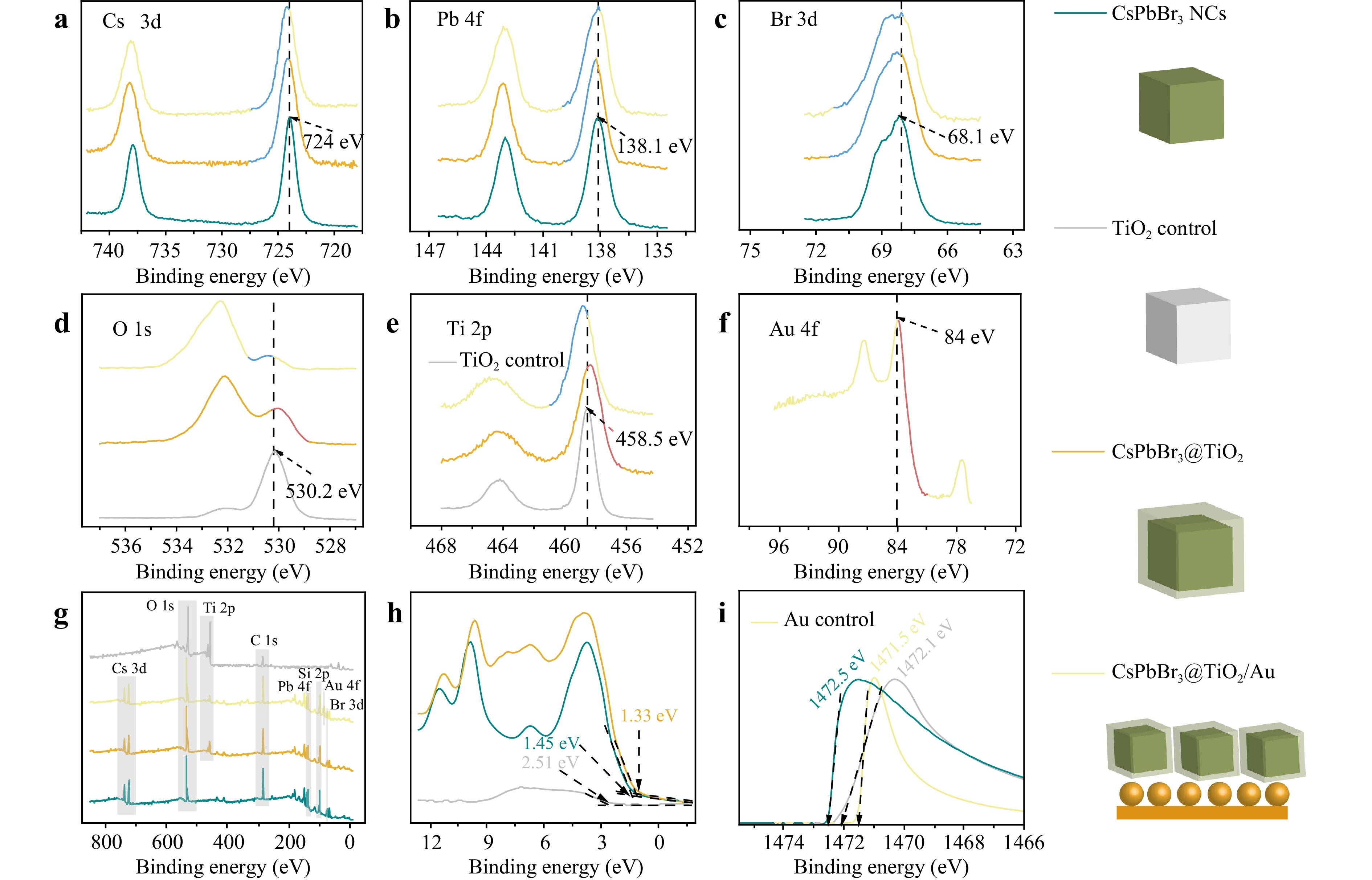
Fig. 3 XPS spectra of TiO2, CsPbBr3 NCs, CsPbBr3@TiO2 core-shell NPs and CsPbBr3@TiO2 core-shell NPs deposited on gold substrate: a Cs 3d; b Pb 4f; c Br 3d; d O 1s; e Ti 2p; f Au 4f; g XPS survey spectrum of CsPbBr3 NCs, TiO2 control, CsPbBr3@TiO2 and CsPbBr3@TiO2/Au; h High-resolution valence band XPS spectra of TiO2, CsPbBr3 NCs as control samples, and CsPbBr3@TiO2; i The secondary electron cut-off in XPS spectra (applied -10 V bias) of TiO2 control, Au control, CsPbBr3 NCs.
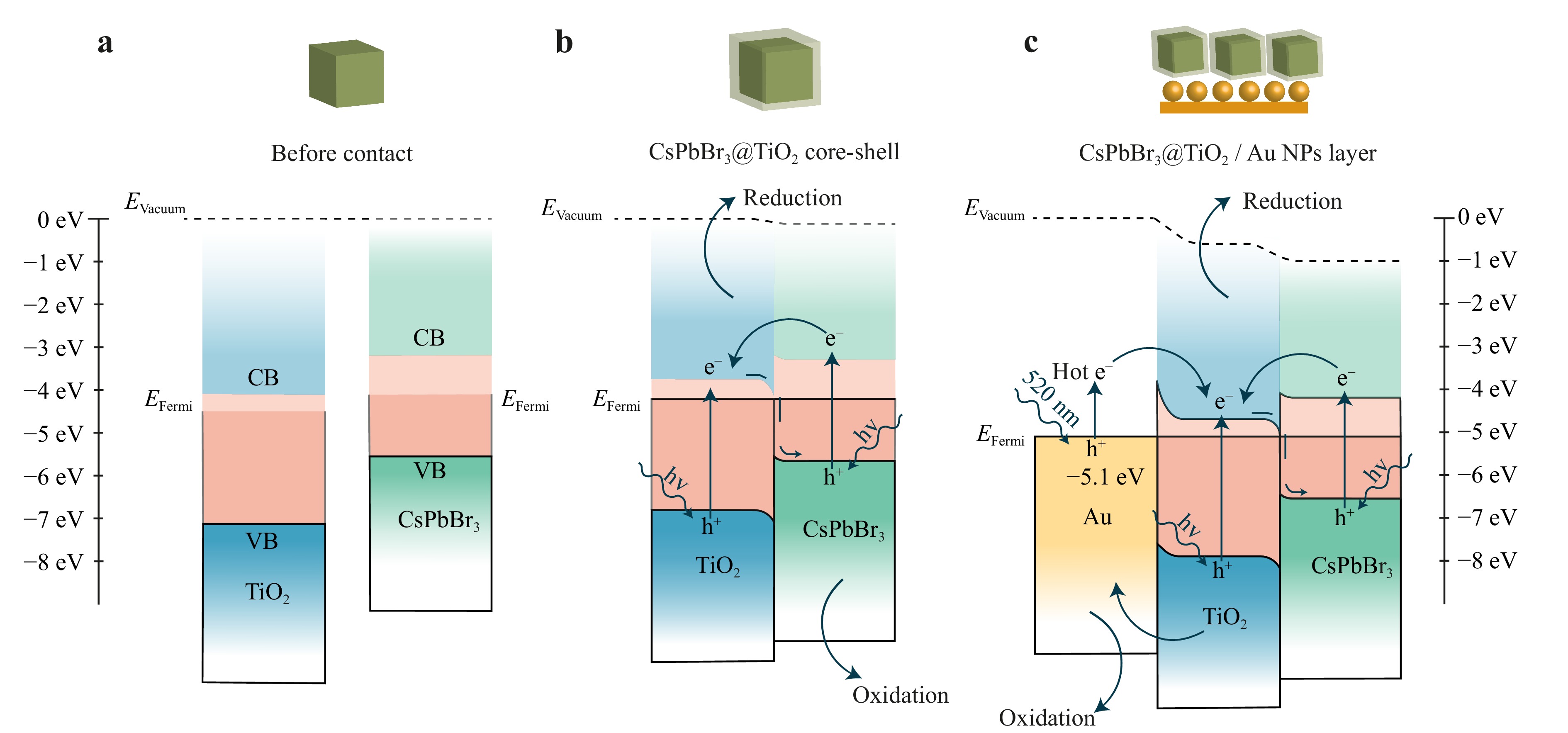
Fig. 4 Band diagrams of a CsPbBr3 and TiO2 before contact, b CsPbBr3@TiO2 core-shell NPs and c CsPbBr3@TiO2 core-shell NPs deposited on Au NPs layer.
The reduced photoelectron signal from the perovskite in CsPbBr3@TiO2 was attributed to the formation of a TiO2 shell, whereas for CsPbBr3@TiO2/Au, it was ascribed to the partial coverage of CsPbBr3@TiO2 with Au NPs (Fig. S1) shows the SEM image, size distribution, and UV-Vis absorption spectra. This finding convincingly demonstrates the successful formation of a shell structure encapsulating the perovskite NCs, as corroborated by HRTEM analysis.
Surface charging effects were ruled out by analyzing the C1s spectra (Fig. S2), which exhibited identical binding energies and FWHM values for all the synthesised samples. Despite the changes in the binding energies of the O1s, Ti2p3, Cs3d5, Pb4f, and Br3d5 photoelectron lines, the stoichiometric ratios for Cs:Pb:Br and Ti:O remained consistent at approximately 1:1:3 and 1:2 (Table S1) for both the CsPbBr3@TiO2 and CsPbBr3@TiO2/Au structures, suggesting no significant stoichiometric deviations in the perovskite core or the TiO2 shell. The observed peak shifts are likely due to interface formation between materials with different band structures26,27.
As shown in Table S2, the photoelectron lines Cs 3d5, Pb 4f7, and Br 3d5 for the CsPbBr3@TiO2 sample shifted towards higher binding energies, whereas the Ti 2p3 and O 1s lines shift to lower values (~0.2 eV). This indicates significant electron charge transfer from the CsPbBr3 NCs to the TiO2 shell. For the CsPbBr3@TiO2/Au sample, the Ti 2p3 and O 1s lines exhibit opposite trends, shifting towards higher binding energies. This suggests the formation of an interface between the TiO2 and Au. The Au 4f7 photoelectron line at 83.8 eV is shifted 0.2 eV below the standard reference, indicating electron transfer from the TiO2 shell to the Au NPs. The Cs 3d5, Pb 4f7, and Br 3d5 lines continue to shift towards higher binding energies, which is consistent with the enhanced charge transfer from the CsPbBr3 core to the TiO2 shell and the formation of a double interface that alters the band alignment.
Notably, the Pb 4f7 peak retains its symmetric shape and exhibits only minor and uniform broadening (FWHM ~1.15–1.18 eV) with no observable shoulders or additional components. Furthermore, no signals corresponding to Pb–O, Pb–OH, or carbonate species are detected in the Pb 4f, O 1s, and C 1s spectra (Fig. 3, Fig. S2). This indicates that the observed shifts are not due to the degradation of the perovskite lattice, rather they arise from interfacial effects and dipole formation at the CsPbBr3/TiO2 and TiO2/Au boundaries. These features underscore the role of Au in tuning the heterojunction structure and facilitating interfacial charge separation, which contributes to enhanced photocatalytic activity.
An equilibrium is established at the interface between semiconductors having different work functions (WF) and Fermi levels through charge transfer. This gives rise to band bending and the formation of depletion regions. These phenomena occur as the system adjusts to align the Fermi levels of the two materials, thereby minimising the energy difference at the interface. The redistribution of charge modifies the local electrostatic potential, resulting in shifting of the positions of the energy bands relative to the vacuum level, which, in turn, alters the binding energies of the emitted photoelectrons.
Although ultraviolet photoelectron spectroscopy (UPS) is widely used to probe the valence band edges and work functions, its application to nanocomposite systems such as CsPbBr3@TiO2/Au is limited because of several critical factors. These materials exhibit strong adsorption of adventitious carbon (Fig. S2) from ambient exposure and possess a rough and developed heterogeneous morphology. Consequently, UPS measurements are highly susceptible to surface contamination, which distorts the secondary electron cutoff and valence band edge, making accurate data acquisition unreliable. Although ion sputtering is often employed to clean surfaces prior to UPS measurements, this step is not suitable in our case because it irreversibly damages the composite structure and alters the interfacial electronic properties. In contrast, XPS with Al Kα 1486.6 eV excitation enables the analysis of subsurface layers up to ~5 nm in depth, directly corresponding to the shell thickness and interfacial region of our heterostructures. Combined with its sufficient energy resolution (~0.01 eV), XPS is a more appropriate technique for assessing interfacial charge redistribution in CsPbBr3@TiO2/Au systems.
Thus, to confirm the presence of interfaces in the CsPbBr3@TiO2 and CsPbBr3@TiO2/Au structures, the valence band maximum (VBM), WF, and bandgap (Eg) were determined using XPS and UV-Vis spectroscopy.
The high-resolution valence band (VB) spectra of the CsPbBr3 NCs, TiO2 control samples, and CsPbBr3@TiO2 structures are shown in (Fig. 3h). Because of the significant overlap between the VB of the CsPbBr3 NCs and TiO2, determining the level shifts in the CsPbBr3@TiO2 structure was challenging. Nevertheless, VBM values of 1.45 eV for NPs and 2.51 eV for TiO2 suggest that the primary contribution to VBM shifts in the CsPbBr3@TiO2 structure comes from the CsPbBr3 NCs. This red shift (~0.12 eV) indicates interface formation between the CsPbBr3 NCs and TiO2, leading to charge redistribution, Fermi level alignment, and band bending. Given the dominant contribution of CsPbBr3 to the VBM of the CsPbBr3@TiO2 system, the perovskite likely acts as an electron donor, with its bands bending downward as follows:
$$ {VBM}_{CsPbBr3}-{VBM}_{CsPbBr3@TiO2}= 0.12\;{\rm{eV}} $$ (1) Using VBM and band gap values, assuming Ef = 0, the Fermi level relative to the conduction band minimum (CBM) is 0.91 eV for CsPbBr3 NPs and 0.46 eV for TiO2, consistent with n-type conductivity in both materials. The work functions of $ {WF}_{CsPbBr3}=4.1\;{\rm{eV}} $ and $ {WF}_{TiO2}= 4.5\;{\rm{eV}} $ (Fig. 3i). These results align with the hypothesis of downward band bending in CsPbBr3 relative to the Fermi level, whereas the TiO2 bands bend upward for Fermi level alignment. The VBM shift of 0.12 eV and a work function difference of 0.4 eV yield a TiO2 band bending estimate, relative to the Fermi level, of
$$ \begin{split}&\Delta \phi -{(VBM}_{CsPbBr3}-{VBM}_{CsPbBr3@TiO2})\\=\;&0.4\;{\rm{eV}}-0.12\;{\rm{eV}}=0.28\;{\rm{eV}} , \\&\Delta \phi ={WF}_{TiO2}-{WF}_{CsPbBr3}=0.4\;{\rm{eV}}\end{split} $$ (2) indicating asymmetric Fermi level shifts within the bandgaps of CsPbBr3 and TiO2 relative to the vacuum level. The Fermi level shift upon forming semiconductor contacts depends on the carrier concentration, as described by
$$ \Delta {E}_{f}=\Delta \phi \times \frac{{N}_{CsPbBr3}}{{N}_{CsPbBr3}+{N}_{TiO2}} $$ (3) where $ {N}_{CsPbBr3} $ and $ {N}_{TiO2} $ are the carrier concentrations in CsPbBr3 and TiO2, respectively.
The band diagrams were constructed based on the obtained VBM, Ef, and WF values (Fig. 4). In the initial stage (before contact), the energy levels of CsPbBr3 and TiO2 are independent, with their conduction bands (CB) and VB separated and their Fermi levels located at different energy positions. After the formation of the CsPbBr3@TiO2 structure, Fermi-level alignment occurs, causing band bending. The CB and VB of CsPbBr3 shift downward by approximately 0.12 eV, while the bands of TiO2 shift upward by about 0.28 eV, creating a barrier height of 0.4 eV. The overall Fermi level of the system is established at approximately −4.22 eV. This band bending leads to the realisation of an “S-scheme” heterojunction, where photoexcited electrons from TiO2 recombine with holes from CsPbBr3 at the interface. Simultaneously, charge separation is effectively maintained, with electrons remaining in CsPbBr3 and holes in TiO2, facilitating the photocatalytic processes.
The use of spherical colloidal gold NPs as plasmonic enhancers has been widely reported because of their well-defined optical properties and ability to promote hot-electron generation28–32. The addition of Au NPs to the CsPbBr3@TiO2 structure creates a second interface at the TiO2/Au boundary, resulting in the formation of a Schottky barrier. Under photoexcitation, hot electrons generated in the Au NPs can overcome this barrier and enter the conduction band of TiO2, where they participate in interfacial redox reactions. Simultaneously, charge separation is further improved owing to the presence of this energy barrier, which suppresses electron backflow and promotes directional carrier flow. Band diagrams (Fig. 4), including the observed energy shifts (0.12 eV for CsPbBr3 and 0.28 eV for TiO2), are consistent with the XPS analysis and confirm the formation of both S-scheme and Schottky interfaces, highlighting the cooperative role of each component in optimizing the charge transfer and in enhancing the photocatalytic performance.
We performed UV-Vis absorption, PL, and TRPL measurements to gain a deeper understanding of the optical properties and recombination processes (Fig. 5). The absorption properties (Fig. 5a, c) of CsPbBr3@TiO2 were higher than those of the pure CsPbBr3 NCs, particularly in the UV region. According to the PL measurements (Fig. 5b), the peaks of the CsPbBr3 NCs of the CsPbBr3@TiO2 NPs correspond to 511 and 514 nm. This red shift can occur for TiO2 coated NPs because of the removal of the lowest fraction of NPs during the purification process. At the same time, a drastic decrease in the PL intensity is observed, suggesting that the catalytic efficiency increased. As the radiation recombination rate decreases, charge separation improves, potentially increasing the amount of charge directed towards the reaction.18 As a result of the excitation mapping of the CsPbBr3 NCs and CsPbBr3@TiO2 NPs, strong quenching of the PL Intensity was observed in the range 265–480 nm. This range covers the excitation range of the UV-Vis lamp used in the catalytic experiments. Fig. 5d, e present the Tauc plots for the CsPbBr3 NCs and CsPbBr3@TiO2 structures (d) and the TiO2 control sample (e). The CsPbBr3@TiO2 structure’s band gap value of 2.36 eV matches that of the CsPbBr3 NCs, indicating that the perovskite structure remains intact after TiO2 shell formation, as confirmed by the HRTEM, XRD, and XPS data.
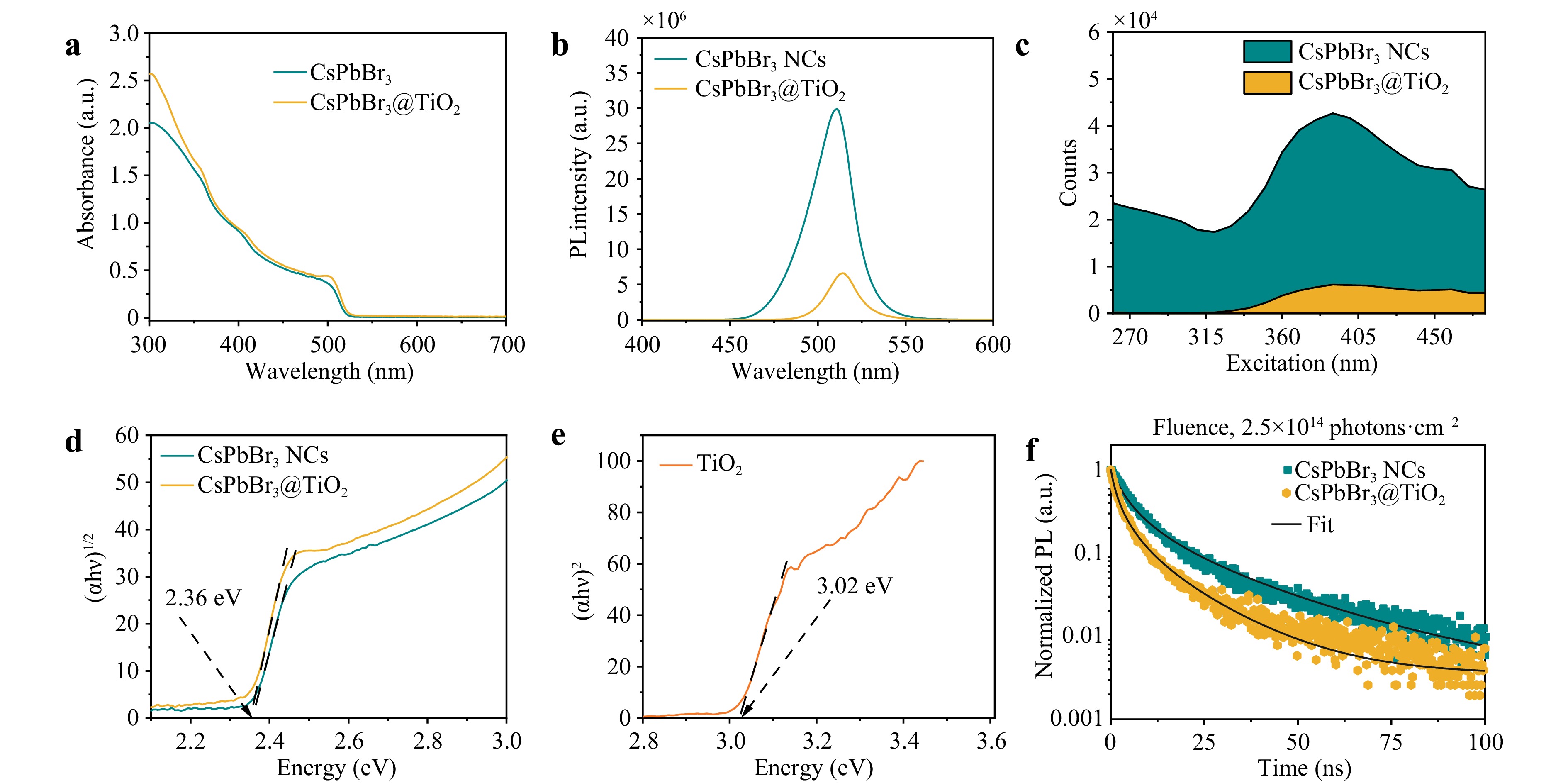
Fig. 5 Optical characterization of CsPbBr3 NCs and CsPbBr3@TiO2 core-shell NPs. a Absorption spectra of CsPbBr3 NCs and CsPbBr3@TiO2 core-shell NPs. b PL spectra of CsPbBr3 NCs and CsPbBr3@TiO2 core-shell NPs. Excitation wavelength corresponds to 375 nm c Excitation mapping of CsPbBr3 NCs and CsPbBr3@TiO2 core-shell NPs. d-e Tauc plots of CsPbBr3 NCs and CsPbBr3@TiO2 NPs. f TRPL measurements of CsPbBr3 NCs and CsPbBr3@TiO2 core-shell NPs. Excitation wavelength corresponds to 375 nm, frequency corresponds to 100 kHz, power corresponds to 6.3 µW.
TRPL spectrophotometry (Fig. 5f) was used to examine the recombination process. During this analysis, we estimated the initial charge carrier density for TiO2 (1.2 × 1017 cm−3) and CsPbBr3 (2.77 × 1017 cm−3) (Table S3), which are necessary for adjusting the energy levels and their bending in the band structures of the diagram (Fig. 5). By substituting the calculated carrier concentration values into (Eq. 2), the Fermi level shifts for each material were determined as approximately 0.11 eV downward for CsPbBr3 and 0.29 eV upward for TiO2. These shifts reflect the redistribution of charge carriers at the interface and are consistent with the VBM positions obtained from the XPS analysis, confirming the alignment of the Fermi levels in the system.
The ABC model (Eq. 4) was used for this calculation because it is suitable for perovskites with low charge carrier concentrations33
$$ -\frac{dn}{dt}=An+B{n}^{2}+C{n}^{3} $$ (4) Term A is responsible for trap-assisted recombination, B for bimolecular recombination, and C for Auger recombination. At low fluence, Auger recombination does not contribute to charge carrier recombination;34 thus, the normalised PL (Eq. 5) decay curve exhibits bimolecular behaviour and can be approximated using the following equation:
$$ n\left(t\right)=\frac{{n}_{0}exp(-At)}{1+\dfrac{{Bn}_{0}}{A}(1-exp(-At\left)\right)}+const $$ (5) where n0 is the maximum charge carrier density.
As mentioned previously, the PL intensity of CsPbBr3@TiO2 is much weaker than that of pristine CsPbBr3 NCs (Fig. 5b), confirming that radiative recombination is suppressed because of the feasibility of charge transfer across the interface of the CsPbBr3@TiO2 heterostructure. This point of view is also confirmed by the results of TRPL (Fig. 5f and Fig. S4) spectrophotometry: The PL lifetime of CsPbBr3 NCs (31.5 ns) is twice that of CsPbBr3@TiO2 (17.4 ns), which once again indicates that part of the radiative recombination is reduced by the addition of TiO2 (1.7 ns) (Table S3-S4). The CsPbBr3@TiO2 NPs exhibited a faster PL decay than the CsPbBr3 NCs, which can be attributed to the increase in carrier migration in this S-scheme heterostructure17. This suggests an improvement in the catalytic activity. Moreover, we observed that the PL lifetime of CsPbBr3@TiO2/Au (12.1 ns) (Table S4) exhibited an even faster decay time than the CsPbBr3@TiO2 NPs, corresponding to the idea that reducing the radiation recombination rate enhances charge separation, thereby improving the catalytic performance of the system. Using the TRPL spectrophotometry results, we calculated the electron transfer rate (Ket = 0.25 × 108 ns−1) using the following equation35:
$$ {K}_{et}=\frac{1}{t(CsPbB{r}_{3}@Ti{O}_{2})}-\frac{1}{t\left(CsPbB{r}_{3}\right)} $$ (6) The value obtained by He et al. corresponds to 5.57 × 108 ns−1. In that study, the major component of the particles was attributed to TiO2 demonstrating a low difference between the well-known TiO2 catalysts35. In contrast, in our study, a lower TiO2 content relative to that of CsPbBr3 resulted in a lower electron transfer rate. However, this difference does not fully reflect the charge-separation ability of the catalyst. In this study, we used a smaller amount of TiO2 to achieve greater stability while still demonstrating the full range of advantages of perovskite-based catalysis, such as tunable reaction energies for various reactions.
To further investigate the role of the heterojunction in the interfacial charge separation and transport, TPC measurements were performed under periodic illumination (Fig. S6). The CsPbBr3@TiO2/Au heterostructure exhibited a markedly enhanced photocurrent response compared to the CsPbBr3@TiO2 and pristine CsPbBr3 NCs, both in amplitude and stability. This enhancement is indicative of more efficient photoinduced charge separation and improved carrier extraction across the dual-interface architecture comprising the CsPbBr3/TiO2 and TiO2/Au junctions.
Complementary EIS measurements (Fig. S7) performed under both dark and illuminated conditions reveal a significant reduction in charge transfer resistance (Rct) upon illumination. In particular, the CsPbBr3@TiO2/Au sample demonstrates the most pronounced decrease in Nyquist semicircle diameter under light exposure, confirming the facilitation of interfacial electron transfer processes in the presence of the plasmonic Au component.
These photoelectrochemical results, together with spectroscopic and structural analyses, provide compelling evidence that the formation of dual heterojunctions and the integration of Au NPs synergistically promote directional charge transport and suppress interfacial recombination, thereby enhancing the overall photoelectrocatalytic performance.
For the CO2 reduction reaction, copper (Cu) foam was used as a reference, along with copper foam impregnated with pristine CsPbBr3 NCs, CsPbBr3@TiO2 core-shell NPs (CT), and CsPbBr3@TiO2 core-shell NPs deposited on Au NPs (CsPbBr3@TiO2/Au, CTA) (Fig. 6a, b). We suggest the use of copper foam because it provides a large surface area for the developed heterostructure designs. Three distinct electrochemical processes were investigated in the electrochemical cell, as detailed in Fig. S8. The results for the copper foam impregnated with CsPbBr3 NCs are not presented, as they are nearly identical to those obtained from catalytic experiments performed on clean copper foam. This similarity is attributed to the low stability of the pristine CsPbBr3 NCs and their fast degradation in KHCO3-water solution. Subsequent experiments demonstrated the stability of the water-resistant CsPbBr3@TiO2 core-shell NPs.
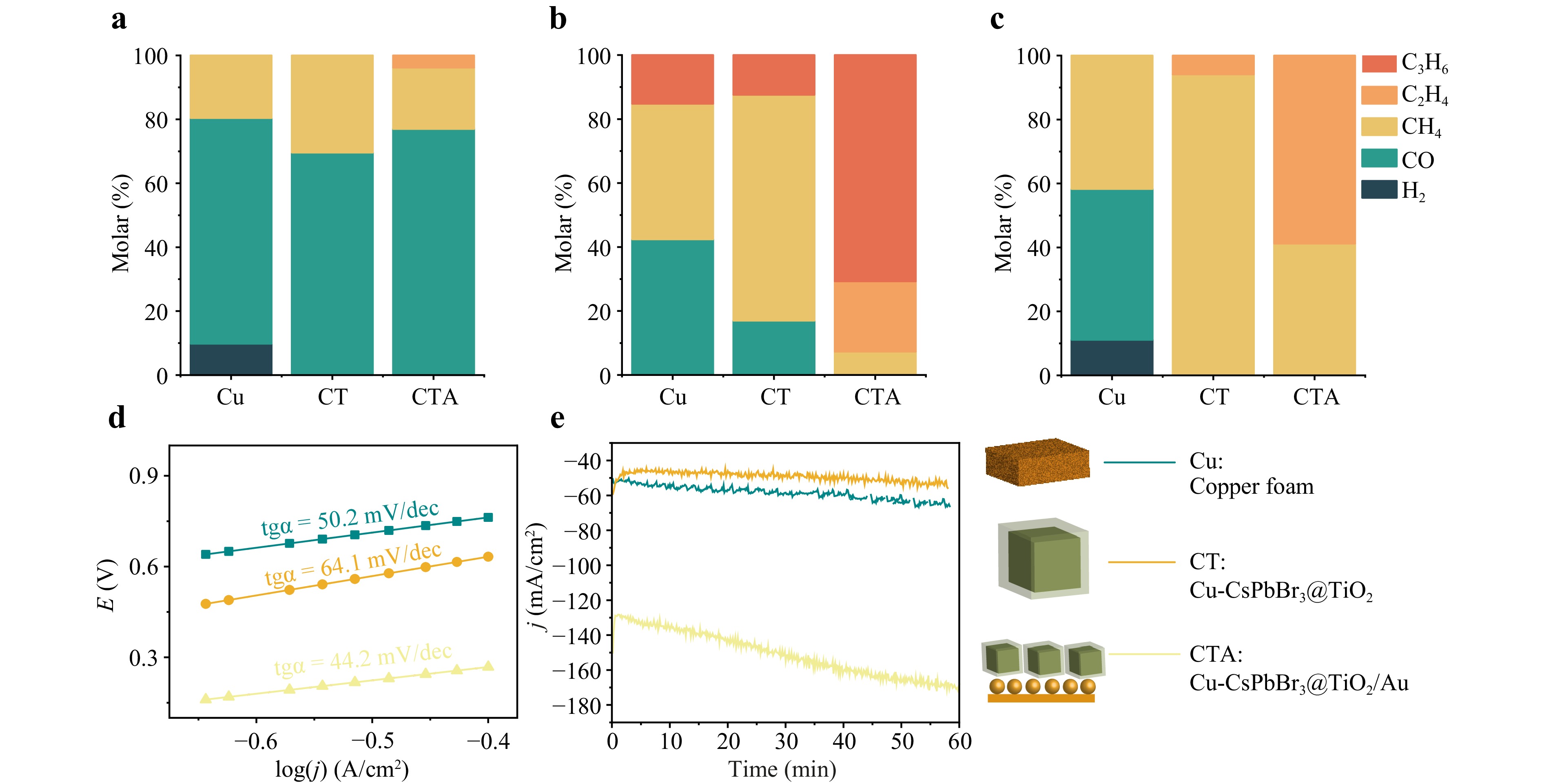
Fig. 6 Catalytic performance of copper foam (Cu) as a reference, copper foam Impregnated with pristine CsPbBr3@TiO2 (CT), and CsPbBr3@TiO2 core-shell NPs deposited on Au NPs (CTA) utilized for a electrocatalysis (EC); b PC; c PEC. d Tafel plot of Cu, CT and CTA for 60 min. e Chronoamperometric measurements of Cu, CT and CTA.
Detailed analysis of the reaction products (over 60 min) using gas chromatography revealed the formation of H2, CO, CH4, C2H4, C3H6 (Fig. 6a-c). O2 was detected only during EC operation and is not included in the PC/PEC product analysis. In this study, we demonstrate the high efficacy of different processes and catalysts in the formation of various products. In electrochemical catalysis, CO was predominantly formed on Cu, CT, and CTA (Fig. 6a), with molar fractions ranging from 70.6% to 77%. Notably, when comparing CT with Cu, there was a 1.5-fold increase in the CH4 production (from 19.6% to 30.4%). Similarly, on comparing CTA with Cu, we observed nearly the same CH4 yield (19.2%), along with the formation of a small amount of C2H4 (3.8%). In contrast, Cu exhibited limited reduction ability, with H2 formation observed at 9.8%. Therefore, in the electrochemical process, we observed a shift in product formation from H2 (on Cu) to CH4 (on CT) and C2H4 (on CTA). In PC (Fig. 6b), 42.4% CO, 42.4% CH4, and 15.2% C3H6 were produced on Cu. For CT, the product distribution shifted significantly with a decrease in CO formation (17%) and an increase in CH4 production (70.6%), accompanied by a small amount of C3H6 (12.4%). For CTA, there was a marked shift towards C2H4 formation (22%) and dominant production of C3H6 (70.7%), with a smaller yield of CH4 (7.3%). Finally, during photoelectrocatalysis (PEC) (Fig. 6c), Cu produced H2 (11.1%), CO (47.2%), and CH4 (41.7%). For CT, we observed a high selectivity for CH4 (94.1%) and a low yield of C2H4 (5.9%). For the CTA, we achieved high yields of CH4 (41.2%) and C2H4 (58.8%). Thus, the most remarkable processes involve the formation of C3H6 on CTA in PC and C2H4 on CTA in PEC. These products are of significant value because of their potential applications as energy sources for chemical syntheses36. Our results indicate that the number of electrons can be influenced, enabling different activation energies involved in the chemical transformations using two different heterostructured designs (CsPbBr3@TiO2 and CsPbBr3@TiO2/Au) and three processes (EC, PC, and PEC).
To evaluate the reaction rate and catalytic stability, we carried out electrochemical measurements such as Tafel plot analysis (Fig. 6d) and chronoamperometry (Fig. 6e). In the Tafel plot (Fig. 6d) for the EC process, we observe Tafel slopes, reflecting the efficiency of charge transfer, of 50.2 mV/dec (Cu), 64.1 mV/dec (CT), and 44.2 mV/dec (CTA). This indicates that the reaction rate decreased in the order CTA > Cu > CT. The lower charge transfer efficiency of CT can be attributed to the presence of TiO2 on the surface of CsPbBr3, because its bandgap is approximately 3.0 eV (Fig. 5). This complicates the charge transfer between the electrode and the material. In contrast, gold-containing CTA showed better charge transfer efficiency. At the same time, the overpotential difference between Cu and CT is approximately 0.15 V, demonstrating the higher catalytic performance of CT37. This difference is much larger for CTA, approximately 0.45 V, further highlighting its enhanced catalytic activity37. Thus, the catalytic efficiency decreased in the following order: CTA > CT > Cu. Moreover, during the EC process, we assessed the stability and efficiency of the materials using chronoamperometry (Fig. 6e). For Cu and CT, we observed an almost constant current density (j) at -50 mA/cm2 for 40 min and 60 min, respectively, indicating that the materials are stable. In contrast, for CTA, we observed an increase in current density from −130 mA/cm2 to −170 mA/cm2 over 60 min, suggesting that the electrochemical reaction becomes more efficient over time38. This improvement can be attributed to catalyst activation, surface modification, or enhanced charge transfer.
Based on the results obtained (Fig. 6a-c), we discuss possible pathways for the chemical transformations. The products of the electro- or photocatalytic conversion of carbon dioxide are usually dominated by CO39, CH440, and CH3OH (methanol),41 with one carbon atom in the structure, called C1 products. Hydrocarbon compounds with two (C2) or more carbon atoms can also be formed, but in most cases in much smaller quantities; nevertheless, they are of considerable interest because they are a crucial chemical feedstock42,43. From this perspective, our results indicate that the conversion of CO2 on the surface of CsPbBr3@TiO2/Au to C3H6 (in PC) and C2H4 (in PEC) exceeded 70% and 58%, respectively, which is of great importance and requires a description of the corresponding mechanisms.
In the literature, two key mechanisms of CO2 conversion with the participation of water in the C1 products are mainly considered: (1) the fast hydrogenation (or formaldehyde) pathway and (2) the fast deoxygenation (or carbene) pathway,44–48 with *COOH and *CHOO as the most likely first intermediates, respectively (* means surface-bound). Most likely, both pathways are realised in practice; however, the predominant role of one is determined by the formation of products and intermediates, which are observed experimentally. The TiO2 surface stabilises the reaction intermediate by forming a chemical bond between CO2 (and HCO3−) and the catalyst, leading to a less negative redox potential47. Thus, CH3OH, along with CH4, is a reaction product of the carbene pathway; therefore, it should be part of the gas mixture obtained as a result of conversion. In the formaldehyde pathway, methanol is an intermediate surface-bound compound formed before the formation of methane; therefore, it was not detected in the reaction products. Because we did not observe the formation of CH3OH, the fast hydrogenation mechanism was chosen as the main mechanism to describe CO2 conversion in our experiments to further propose, on this basis, an idea of how the formation of C2 products occurs. Notably, the formaldehyde pathway can be accomplished by forming CO or formic acid (HCOOH). HCOOH formation is thermodynamically more favourable, while the carbon monoxide formation is kinetically more favourable49. Because we observed the formation of CO in our experiments, the pathway involved was chosen for further discussion (Fig. 7).
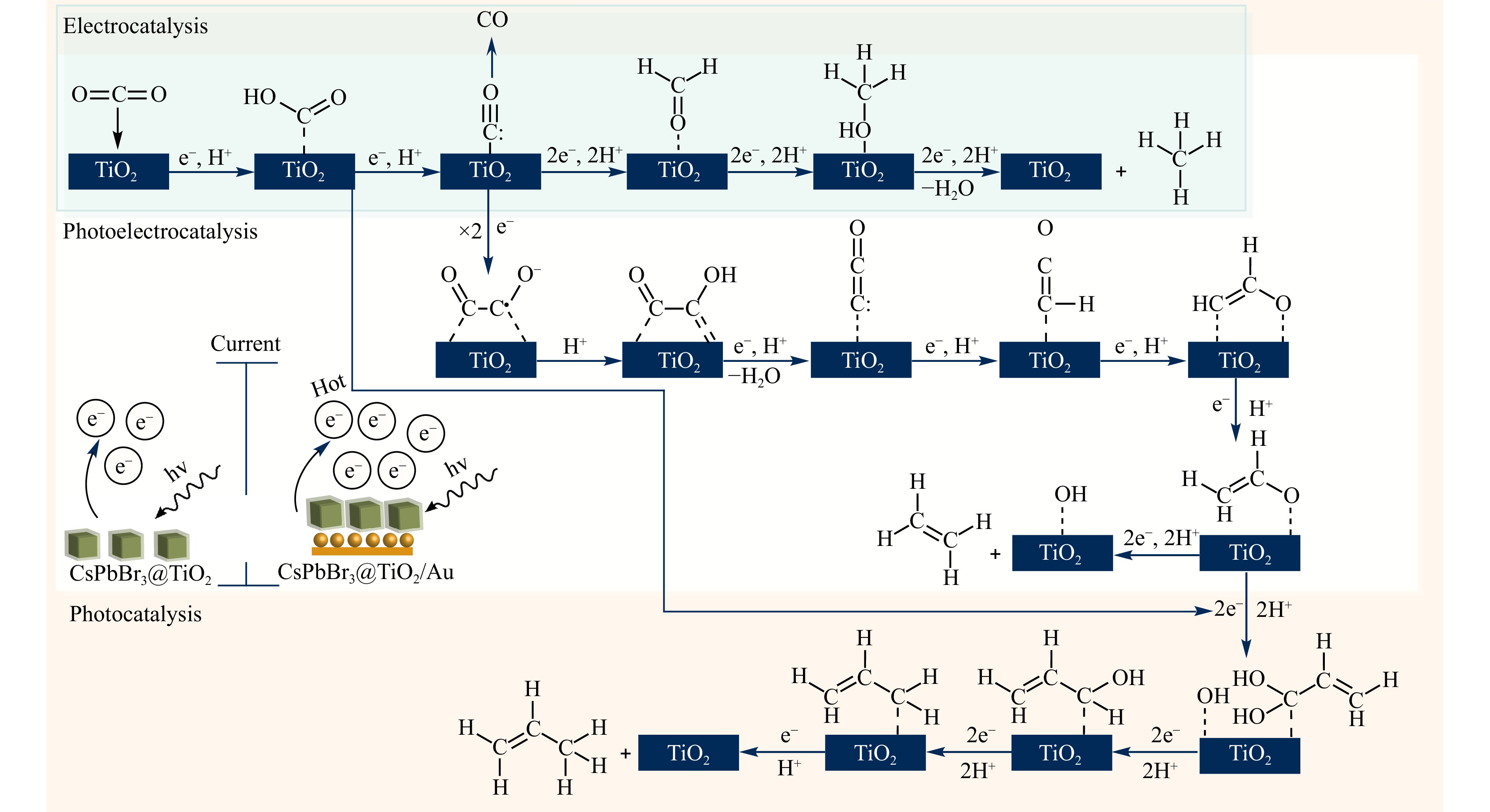
Fig. 7 The proposed scheme of chemical transformations on the surface of CsPbBr3@TiO2 and CsPbBr3@TiO2/Au during CO2 conversion in the three considered catalytic processes with the predominant formation of the corresponding products. The schematic inset illustrates the underlying reason for the formation of different products when using CsPbBr3@TiO2 versus CsPbBr3@TiO2/Au. This distinction arises from the difference in the number of electrons involved in the reaction, ultimately determining the product distribution.
The next step was to move from the mechanism of C1 product formation to the C2 product formation. It has been previously shown that CO2 conversion can yield ethylene50, ethane51, and acetic acid52 with a relatively low selectivity of less than 50%. The mechanism of C2 product formation is assumed to be based on a slow multi-electron reduction and a carbon-carbon (C-C) coupling driven by the high energy generated by illumination53. It has been shown that the binding of two CO molecules occurs through the interaction of carbon atoms with metal atoms with increased electron density13,14. Accordingly, the reaction pathway proposed by Ji et al.54 was used as a reference to explain the formation of C2H4 from CO2 on the CsPbBr3@TiO2/Au surface. In our study, this pathway was further refined by clearly distinguishing between the photocatalytic, electrocatalytic, and photoelectrocatalytic processes (Fig. 7). We did not include all intermediate steps in the reaction scheme, indicating only those steps demonstrating fundamental importance for understanding the reasons for obtaining the observed products.
Summarising the mechanisms of CO2 conversion briefly described above, the following conclusions can be drawn: In all the three catalytic processes considered, the first step was the deoxygenation of CO2 with the formation of CO bound to the TiO2 surface. In the case of EC, a major portion of carbon monoxide molecules is desorbed, and the remaining part participates in the formation of CH4 and, to a lesser extent, in that of C2H4 (Fig. 7). In PEC, most CO molecules are coupled to form COCO compounds and the remaining molecules are converted into methane molecules along the formaldehyde pathway. The formation of COCO dimers was facilitated by excess electrons, which was supported by the proposed heterostructured Schottky/S-scheme of CsPbBr3@TiO2/Au (Fig. 4). These dimers were predominantly converted into C2H4. This interpretation is further supported by the EPR data (Fig. S3), which reveal predominant •OH radical formation, indicative of a high oxidative potential and efficient charge separation. Notably, the EPR spectra of the Au-containing system show a lower relative contribution from O2−• adducts compared to that of the non-Au sample, suggesting that more photogenerated electrons are actively involved in the reduction reactions55. This increased electron availability in the CTA system correlates with the observed shift in the product distribution towards multielectron C2 products. Based on the band-edge positions19, the observed redox behaviour is consistent with the S-scheme mechanism.
Based on recent studies, it can be concluded that propylene is obtained from the coupling of the vinyl alcohol intermediates *C2 and *C1 (and/or *C1') sorbed on the surface of TiO242. These studies have shown that allyl alcohol (the product of the coupling of the aforementioned intermediates, *C1 and *C2) is responsible for the formation of propylene.
-
In this study, we demonstrated a novel strategy for CO2 conversion by integrating perovskite-based photoelectrocatalysis with localised surface plasmon resonance enhancement. To overcome the problem of lead halide perovskite instability in water, we fabricated stable CsPbBr3@TiO2 core-shell NPs and investigated their structural and optical properties. Further, we prepared layered materials based on these NPs and combined them with Au NPs. Furthermore, we explored three distinct catalytic processes — electrocatalytic, photocatalytic, and photoelectrocatalytic — to tune the selectivity of CO2 reduction to a range of valuable products, including H2, CO, CH4, C2H4, and C3H6.
Comprehensive structural and optical analyses of the CsPbBr3@TiO2 core-shell NPs revealed successful synthesis and effective coating of TiO2 on the CsPbBr3 NPs. Through the use of TEM, SEM, XRD, and XPS the formation of high-quality NPs was confirmed and provided detailed insights into their structural and elemental compositions. Moreover, based on XPS and optical measurements (UV-Vis absorption, PL spectroscopy, and TRPL spectrophotometry), we described the electronic band structure necessary for understanding the charge dynamics. The analysed charge dynamics demonstrated excellent enhancements in charge separation, indicating a promising path for improving catalytic efficiency.
Using CsPbBr3@TiO2 and CsPbBr3@TiO2/Au heterostructured designs for electrocatalysis, photocatalysis, and photoelectrocatalysis, we achieved high selectivity in the CO2 reduction reaction. By controlling the selectivity, we demonstrated that CsPbBr3@TiO2 NPs and CsPbBr3@TiO2/Au heterostructures can be tuned to produce valuable product varieties. Notably, our findings highlight the potential of perovskite-based catalysts with Cu foam as a reference material in electrocatalytic experiments. The introduction of Au into the CsPbBr3@TiO2/Au system further enhanced its catalytic performance, demonstrating the beneficial role of localised surface plasmon resonance enhancement in the photoelectrocatalytic process. Under various catalytic conditions, the reaction products shifted towards higher-order hydrocarbons such as C2H4 and C3H6, which are of significant interest for chemical synthesis as energy sources.
In terms of catalytic stability, chronoamperometry experiments revealed that both CsPbBr3@TiO2 core-shell NPs and CsPbBr3@TiO2/Au heterostructures exhibit high stability over extended periods of electrochemical measurements. These findings provide a strong foundation for the future development of stable, efficient, and scalable photoelectrocatalytic systems for sustainable CO2 conversion.
-
This study was financially supported by the National Natural Science Foundation of China (Project No.62350610272). S.V.M acknowledges the Department of Science and Technology of Shandong Province (Grant KY0020240040) and the Priority 2030 Federal Academic Leadership Program. P.O.K. acknowledges the support from the Ministry of Science and Higher Education of the Russian Federation (Project No. FSRZ 2023-0006). Fig. 1 was drawn using pictures from Servier Medical Art. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License.
Heterostructured perovskite nanocrystals for water stable plasmon-enhanced photoelectrocatalysis
- Light: Advanced Manufacturing , Article number: (2025)
- Received: 30 December 2024
- Revised: 20 October 2025
- Accepted: 20 October 2025 Published online: 25 November 2025
doi: https://doi.org/10.37188/lam.2025.080
Abstract: Perovskite photoelectrocatalysis enables solar-driven conversion of CO2 to value-added chemicals, but instability in water and insufficient C–C coupling still constrain performance. Herein, we present a synergistic approach for aqueous-phase CO2 conversion that combines perovskite-based photoelectrocatalysis with localized surface plasmon resonance (LSPR) enhancement. To address the inherent instability of lead-halide perovskites, we developed a modified hot-injection route that enables the in situ formation of water-stable CsPbBr3@TiO2 core–shell nanoparticles. Titanium butoxide and water were introduced after Cs-oleate injection, enabling controlled TiO2 shell growth without post-treatment. Electron microscopy, XRD, and XPS confirm the core–shell architecture, while optical/electrical probes indicate efficient charge separation across the CsPbBr3/TiO2 junction. Subsequently, we investigated electrocatalytic, photocatalytic, and photoelectrochemical carbon dioxide reduction (CO2RR) on CsPbBr3@TiO2/Au and CsPbBr3@TiO2. Gas chromatography revealed tunable product selectivity, yielding H2, CO, CH4, and multicarbon (C2, C3) products including C2H4 (ethylene) and C3H6 (propene). Our main findings indicate that the CsPbBr3@TiO2/Au exhibits high selectivity toward C3 (propene) in photocatalysis and C2 (ethylene) in photoelectrochemistry, reaching up to 70% and 58%, respectively. These results highlight perovskite heterostructures as a viable platform for efficient CO2 utilization and the sustainable production of value-added C2/C3 chemicals.
Research Summary
Halide Perovskite heterostructures enable efficient valuable chemicals obtain
Rising carbon dioxide levels demand new ways to transform this greenhouse gas into useful products. Usually halide perovskites die when they contact with water. Researchers have developed water-stable perovskite–titania nanoparticles that, when combined with gold, create a powerful platform for light-driven CO2 conversion. The combination of the unique design of these particles with gold and the use of different stimuli, such as light or electricity, enables the production of valuable chemicals. The main result was the selective conversion to valuable chemicals such as ethylene and propene, with selectivities above 58% and 70%, respectively, which is considered high. This advance demonstrates how perovskite-based heterostructures can be engineered to not only tackle CO2 emissions but also generate high-value feedstocks for the chemical industry.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article′s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article′s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.

 DownLoad:
DownLoad: